

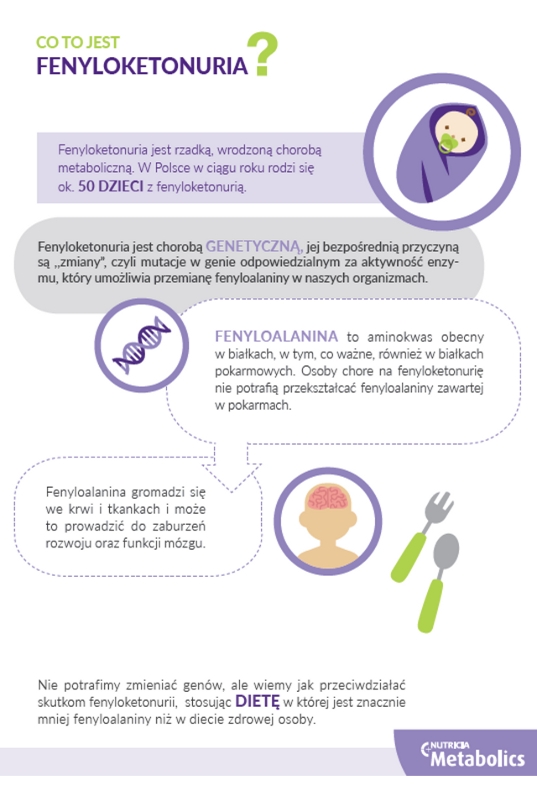
Fenyloketonuria (PKU) jest wrodzoną chorobą metaboliczną, dziedziczoną od obojga rodziców, którzy nie mają objawów choroby, lecz są nosicielami zmienionego genu. W Polsce jeden na siedem – osiem tysięcy noworodków rodzi się z fenyloketonurią. Oznacza to, że każdego roku przybywa w naszym kraju ok. 50 dzieci z PKU. To wrodzona wada metabolizmu, która wymaga przestrzegania przez całe życie ścisłej diety ubogofenyloalaninowej. Jest to codzienne wyzwanie, ale konsekwentnie realizowane daje szansę na normalne życie.
Niebezpieczna fenyloalanina
– Przyczyną fenyloketonurii jest częściowy deficyt lub całkowity brak aktywności produkowanego w wątrobie enzymu hydroksylazy fenyloalaniny. Odpowiada on za przemianę jednego aminokwasu – fenyloalaniny, do innego aminokwasu- tyrozyny. Aminokwasy to małe „cegiełki”, które są składnikami wszystkich białek, niezbędnych do prawidłowego funkcjonowania wszystkich organizmów żywych, w tym człowieka. W rezultacie zablokowania przemiany fenyloalaniny, jej nadmiar gromadzi się we krwi i wywiera szkodliwe działanie na tkanki organizmu, a szczególnie na układ nerwowy. Głównym objawem klinicznym nieleczonej fenyloketonurii jest postępująca niepełnosprawność intelektualna, zwykle w stopniu znacznym. Nieleczony chory z PKU nie jest samodzielny, nie nabywa umiejętności właściwych dla określonego etapu rozwoju, często traci nabyte już umiejętności. W skrajnych przypadkach nie chodzi, nie mówi, nie je samodzielnie. Pojawiają się też inne objawy neurologiczne, w tym padaczka – tłumaczy doktor Dorota Korycińska-Chaaban z Poradni Chorób Metabolicznych Instytutu Matki i Dziecka w Warszawie.
Szybka diagnoza noworodka szansą na zdrowie
Chociaż aktualnie nie jest stosowane leczenie przyczynowe PKU, istnieje skuteczna metoda zapobiegania wystąpieniu objawów uszkodzenia układu nerwowego – to specjalna dieta ubogofenyloalaninowa, jedyna dostępna w Polsce metoda leczenia fenyloketonurii. Warunkiem sukcesu jest rozpoczęcie leczenia dietą z ograniczeniem podaży fenyloalaniny przed wystąpieniem objawów klinicznych choroby. Jest to możliwe dzięki Programowi Badań Przesiewowych Noworodków finansowanych przez Ministerstwo Zdrowia. W trzeciej dobie życia od noworodka pobiera się krew na specjalną bibułę, a następnie przesyła się ją ze szpitala do Zakładu Badań Przesiewowych. Z suchej kropli krwi na bibule wykonuje się badania w kierunku licznych wrodzonych wad metabolizmu, w tym fenyloketonurii. Stwierdzenie podwyższonego stężenia fenyloalaniny we krwi dziecka wymaga dalszej diagnostyki w ośrodkach specjalistycznych. W Warszawie jest to Klinika Wrodzonych Wad Metabolizmu i Pediatrii w Instytucie Matki i Dziecka.
– Rozpoznanie fenyloketonurii w pierwszych dniach życia oraz wdrożenie odpowiedniego leczenia zapewnia chorym rozwój intelektualny podobny do ich zdrowych rówieśników.
Prawidłowo i systematycznie leczeni pacjenci z PKU uczęszczają do żłobków, przedszkoli, kończą szkoły, studia, pracują w wybranych zawodach, zdobywają tytuły naukowe, zakładają rodziny i rodzą zdrowe dzieci. Wysokie stężenia fenyloalaniny wywierają toksyczne działanie na mózg w każdym wieku, a wysokie stężenia fenyloalaniny we krwi ciężarnej kobiety z PKU szkodliwie działają na rozwijający się płód, powodując zespół wad wrodzonych, nazywany Zespołem Fenyloketonurii Matczynej. Dlatego też leczenie dietą ubofenyloalaninową jest konieczne przez całe życie. To bardzo ważne, aby każdy chory z PKU, w każdym wieku, był objęty opieką Poradni Metabolicznej. Szczególnie dotyczy to małych dzieci, młodzieży i kobiet w wieku rozrodczym – mówi doktor Dorota Korycińska-Chaaban.
Rygorystyczna dieta pełna wyrzeczeń
Dieta eliminacyjna stosowana w leczeniu fenyloketonurii jest dietą pokrywającą zapotrzebowanie na wszystkie makro- i mikroelementy, witaminy oraz energię tak jak u osób zdrowych, jednakże o obniżonej ilości fenyloalaniny oraz zwiększonej zawartości białka.
Podstawowe zalecenia diety ubogofenyloalaninowej dotyczą następujących składników: fenyloalaniny, tyrozyny, białka i energii. Produkty wysokobiałkowe, mające duży udział w diecie osób zdrowych takie jak: mięso i produkty mięsne, nabiał, ryby, jaja, rośliny strączkowe, produkty mączne, orzechy, kakao, czekolada, muszą być wykluczone z diety pacjentów z PKU.

– Codzienne stosowanie diety wymaga szczegółowego zaplanowania posiłków chorego, dokładnego ważenia każdego produktu i obliczania spożywanej ilości fenyloalaniny, białka oraz kalorii. Chory może otrzymać z posiłkami ściśle określoną ilość fenyloalaniny, zgodnie z aktualną tolerancją. Na szczęście w obliczeniach pomocne są dostępne specjalne kalkulatory PKU. Białko, niezbędne organizmowi do odpowiedniego rozwoju, chorzy z PKU otrzymują w postaci specjalnych preparatów – mieszanin syntetycznych aminokwasów, pozbawionych fenyloalaniny, będących równoważnikiem białka. Szeroka gama produktów niskobiałkowych dostępna jest w coraz większej ilości zwykłych sklepów, co zdecydowanie ułatwia komponowanie atrakcyjnej i urozmaiconej diety – komentuje dr n. med. Joanna Żółkowska, dietetyk Poradni Chorób Metabolicznych Instytutu Matki i Dziecka w Warszawie.
– Rzadko mówię o tym, że jestem chory – dieta PKU to po prostu część mojego stylu życia. Intuicyjnie wiem, kiedy i jak często powinienem przygotowywać posiłki – kontroluję swoje wyniki, które są dla mnie punktem odniesienia w planowaniu jadłospisu. Pomimo ścisłych rygorów i dyscypliny, moja kuchnia to czysta improwizacja i niekonwencjonalne łączenie różnych składników. Po wielu latach dbania o odżywianie, picie preparatu staje się też normalnym odruchem – zupełnie jak oddychanie – dodaje Bartłomiej Boroń, pacjent chory na fenyloketonurię.
#Fenymenalni
Z myślą o codziennym wsparciu osób z fenyloketonurią już ponad 7 lat temu powstała platforma PKU Connect, dzięki której osoby związane z PKU mogą czerpać wiedzę, pomysły na posiłki, a także pozytywną energię i motywację do działania. To nie tylko przestrzeń wirtualna. Pacjenci poznają się również osobiście podczas warsztatów organizowanych przez Nutricia Metabolics, markę działającą w ramach firmy Nutricia Polska. Cyklicznie wydawany jest też drukowany magazyn, który tworzą eksperci i #Fenymenalni pacjenci.
Więcej informacji znaleźć można pod adresem: https://fenymenalni.pkuconnect.pl/.